Глава
59. Врожденные коллагенозы
John Keith Jenkins, M.D.
Глава
60. Врожденные дефекты метаболизма соединительной ткани
John Keith Jenkins, M.D.
Глава
61. Болезни накопления
Markjarek, M.D., FASR
Глава
62. Ревматологические проявления синдромов первичного иммунодефицита
MarkMalyak,M.D.
Глава
63. Дисплазии костей и суставов
Edmund H. Hornstein,
D.O.
X. Наследственные и
врожденные нарушения метаболизма при ревматических синдромах
Закон наследования объясняет, почему
во всем что есть в вашем ребенке плохого, виноват другой родитель.
Неизвестный автор
ГЛАВА 59. ВРОЖДЕННЫЕ
КОЛЛАГЕНОЗЫ
John Keith Jenkins, M. D.
1. Какие основные типы
коллагена присущи человеческому организму? В каких органах и тканях они
находятся?
Коллаген I типа составляет
60-90 % сухой массы кожи, связок и деминерализован-ной кости. Коллаген
II типа составляет > 50 % сухой массы суставных хрящей; содержится в значительном
количестве в стекловидном теле, пульпозном ядре позвоночника, хрящах носа
и ушных раковин. III тип коллагена обнаруживают в кровеносных сосудах и
тканях, в которых есть I тип коллагена (за исключением кости). IV тип встречается
в большинстве видов базальных мембран. VII тип является также коллагеном
базальных мембран и присутствует в коже.
2. Опишите основные черты
синтеза коллагена.
Синтез коллагена — сложный
процесс, который происходит при нормальной экспрессии генов, отвечающих
за его синтез. Молекулы его должны выполнять определенные функции. Ядрообразующий
рост нового коллагенового волокна требует не только правильной первичной
структуры, но и посттрансляционных изменений молекул проколлагена. Например,
коллаген I типа образован из двух молекул прокол-лагена I типа а,-цепи
и одной молекулы I типа а2-цепи. Три цепи соединяются вместе
с одного конца, образуя тройную спираль (ядрообразующий рост) после соответствующего
посттрансляционного изменения пролиновых остатков. Четыре фибриллы соединяются
между собой в шахматном порядке и формируют большое волокно.
3. Каким образом аномалии
синтеза коллагена приводят к клиническим проявлениям болезни?
1. При мутациях генов, отвечающих
за синтез коллагена, образуется аномальный тип коллагена. Наличие этого
аномального коллагена в органе приводит к его поражению.
2. Мутации, которые только
снижают продукцию определенного типа коллагена, сопровождаются менее выраженными
клиническими формами поражения (фенотипами), чем те мутации, которые вызывают
высокий уровень экспрессии структурно аномальных типов коллагена.
3. Принцип ядрообразующего
роста объясняет, почему наличие структурно аномальных молекул коллагена
приводит к выраженным и потенциально летальным клиническим исходам.
4. Разнообразные замены аминокислотных
остатков в одних и тех же генах коллагена способны нарушать функции молекул
и вызывать не одинаковые, но перекрывающиеся клинические синдромы.
4. Что такое osteogenesis
imperfecta? Какие органы при этом поражаются и почему?
Osteogenesis imperfecta
(OI)
— заболевание, известное также под названием "врожденная ломкость костей".
В действительности это группа заболеваний со схожими клиническими проявлениями
(хрупкие кости и голубые склеры) различной степени тяжести, с предположительно
одной этиологией, но разными типами наследования. В последних исследованиях
получены удивительные результаты, свидетельствующие, что более чем у 90
% всех больных OI наблюдаются мутации в одном из двух генов, кодирующих
продукцию коллагена I типа. Эти аномалии коллагена вызывают остеопению
и хрупкость кости и, как следствие, частые переломы. Склера при снижении
содержания в ней коллагена приобретает голубоватый оттенок, поскольку через
нее просвечивают внутренние среды глаза.
5. Мутации в гене коллагена
I типа приводят к хрупкости кости. Имеются ли дефекты молекулы коллагена
при остеопорозе?
Недавно установлено, что
дефекты молекулы коллагена являются причиной некоторых форм семейного остеопороза
в Европе и США. Некоторые исследователи полагают, что женщины группы риска
по остеопорозу могут иметь пока не идентифицированную аномалию коллагена,
которая предрасполагает к возникновению остеопороза "недоношенных".
6. Почему дефект коллагена
I типа приводит к возникновению болезни хрупкости кости у одного пациента
и семейному остеопорозу у другого?
Коллаген — сложная молекула,
содержащая множество доменов с различной функцией. Клинические проявления
аномалий молекулы изменяются в зависимости от нарушенной структуры или
функции. В последние годы обнаружились частичные совпадения в клинических
картинах нетяжелой формы osteogenesis imperfecta и синдромов остеопороза,
а также между osteogenesis imperfecta и синдромами Элерса-Дан-лоса.
Идентификация молекулярного дефекта определенного типа коллагена не обязательно
приводит к постановке точного клинического диагноза. Эти заболевания подтверждают
правило Орвеллиана (Orwellian) — некоторые типы коллагена больше похожи
друг на друга, чем другие. Первое следствие этого правила — необходимо
изучать клинические синдромы заболевания.
7. Что такое классификация
Силленса (Sillence) osteogenesis imperfecta?
Согласно классификации Силленса,
клинические виды osteogenesis imperfecta сгруппированы в четыре
категории по степени тяжести. Множество мутаций могут лежать в основе каждой
категории этой классификации.
Тип IOI (легкий). У больных
отмечается хрупкость костей с умеренным количеством переломов (приводящих
к червеобразному искривлению длинных трубчатых костей), голубые склеры
и малый рост.
Тип II OI (обычно
летальный). У больных отмечают множество переломов еще внутриутробно. Высокая
внутриутробная смертность или смертность в периоде новорожденное™.
Тип III OI (тяжелый,
деформирующий). У больных отмечают многочисленные деформации скелета, ломкость
костей, сколиоз и разболтанность суставов с большим количеством переломов.
Склеры пациентов серые или голубые, плотность костной ткани снижается (остеопения).
Обычно больные не наблюдаются и лечатся стационарно.
Тип IV OI (средней
степени тяжести). У больных этой группы также отмечается большое число
переломов, червеобразное искривление длинных трубчатых костей, нормальные
(белые) или серые склеры, снижение плотности костной ткани и деформация
костей; они могут наблюдаться и лечиться амбулаторно.
Тип III OI наследуется аутосомно-рецессивно,
типы I, II и IV — аутосомно-доми-нантно. Аномалии тканей зубов отмечаются
при I, III, IV типах, глухота — при I и IV.
8. Опишите клинические
проявления синдромов Элерса-Данлоса.
Синдромами Элерса-Данлоса
называют группу редких заболеваний, поражающих суставы и кожу, характеризующихся
повышенной
амплитудой движений в суставах, повышенной ранимостью и "сверхрастяжимостью"
кожи. Отмечаются в зависимости от типа синдрома большие различия в
степени поражения суставов, кожи и внутренних органов. Выделяют тип I (gravis)
и
тип II (mitis) по характерным поражениям кожи и суставов.
9. Каково значение повышенной
амплитуды движений в суставах и "сверхрастяжимости" кожи?
Повышенная амплитуда движений
в суставах уменьшает стабильность сустава и увеличивает частоту вывихов.
Разболтанность суставов выражается в виде врожденного вывиха бедра и привычных
вывихов в дальнейшей жизни. При рецидивирующих вывихах сустава и кровотечении
в его полость может возникнуть гемартроз. Кожа таких больных плохо заживает
при мелких ранениях, и из-за ее непрочности происходит прорезывание хирургических
швов.
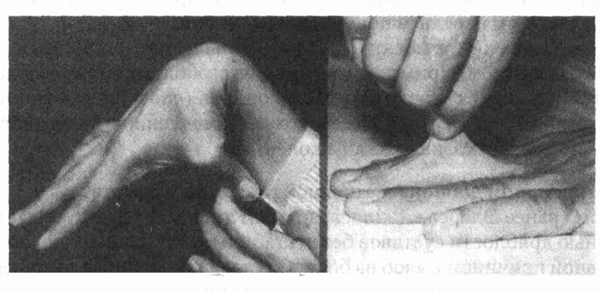
Больной с синдромом Элерса-Данлоса
демонстрирует переразгибание суставов и сверхрастяжимость кожи. (Из: Clinical
Slide Collection on the Rheumatic Diseases. Atlanta, American College of
Rheumatology, 1991; с разрешения.)
10. Как наследуется синдром
Элерса-Данлоса?
Генетический дефект известен
только при нескольких типах этого синдрома. В большинстве случаев наследование
происходит по аутосомно-доминантному типу и сопровождается уменьшением
количества или, иногда, изменением структуры коллагена. Исключение составляет
тип VI синдрома Элерса-Данлоса (окуло-сколиотический), при котором дефект
лизилгидроксилазы наследуется рецессивно.
Этот витамин С-зависимый
фермент необходим для образования гидроксилизина, который участвует в формировании
перекрестносвязанного коллагена. Кроме разболтанности суставов и "сверхрастяжимости"
кожи при этом типе заболевания обнаруживаются сколиоз и грыжа глазного
яблока, хотя в основе лежит только дефект коллагена.
11. Что такое сосудистая
форма синдрома Элерса-Данлоса?
Синдром Элерса-Данлоса IV
типа (сосудистый тип) характеризуется нарушением продукции коллагена III
типа, который находится в стенках кровеносных сосудов. Поражения суставов
и кожи менее выражены, чем при типах I и II, но разрывы сосудов или стенки
кишки могут иметь самые драматические последствия вплоть до летального
исхода Дефект коллагена III типа является самой частой причиной ранних
аневризм сердечно-сосудистой системы (аневризма брюшной аорты) и сосудов
мозга (проксимальнее виллизиева круга) Такие редкие заболевания, как синдром
Элерса-Данлоса и osteogenesis imperfecta, могут быть просто фенотипическими
проявлениями мутаций структурных белков, что проявляется ранним возникновением
болезней костей, суставов и сосудов.
12. Как определить разболтанность
суставов?
Существует шесть простых
тестов, которые проводятся во время физикального обследования. 1 Переразгибание
в коленном суставе более чем на 10°.
2. Переразгибание в локтевом
суставе более чем на 10°.
3. Разгибание первого пальца
кисти до касания передней поверхности предплечья.
4. Разгибание пальцев кисти,
когда ось пальцев становится параллельной оси предплечья.
5. Сгибание туловища со свободным
касанием ладонями пола.
6. Дорсальное сгибание стопы
более чем на 20° от прямого угла между дорсальной поверхностью стопы и
передней поверхностью голени.
Амплитуда движений в мелких
суставах больше, чем в крупных, и она может уменьшаться с возрастом. Примерами
признаков разболтанности/сверхрастяжимос-ти других органов могут служить
симптом Горлина (способность коснуться кончиком языка кончика носа) и дряблость
кожи (замедленное расправление кожной складки и пергаментообразная кожа
на коленях).
13. Что такое синдром
доброкачественной гипермобильности?
Этот синдром (ранее Элерса-Данлоса
синдром типа III) характеризуется неодинаковой степенью дряблости суставов
без нестабильности или неподвижности. Он является основной причиной жалоб
на боли в суставах (коленного, тазобедренного, суставов рук), имеет тенденцию
передаваться по наследству; часто отмечается растяжение связок голеностопного
и лучезапястного суставов. Среди больных преобладают молодые девушки. При
этом синдроме обнаруживается выпот в полость сустава. Данный синдром диагностируется
по наличию по крайней мере трех из шести признаков сверхрастяжимости суставов.
14. Какая система органов
первично поражается при синдроме Марфана?
Глаза, опорно-двигательный
аппарат, сосудистая система, но могут наблюдаться заболевания кожи и дыхательной
системы. У всех пациентов выявляется дефект в гене фибриллина. Наследуется
синдром Марфана по аутосомно-доминантному типу.
Встречаемость — более 6 случаев
на 100 000 человек. Долгое время считалось, что его причиной является нарушение
синтеза эластина в специализированной соединительной ткани, имеющейся в
большом количестве в связках и обеспечивающей эластичность. Однако позднее
не было выявлено никаких аномалий эластина. Фибриллин, обнаруженный в рыхлой
соединительной ткани, является компонентом внеклеточных микрофибрилл, которые
формируют субструктуру эластина. Пораженные органы при синдроме Марфана
— это те, в которых эластические волокна (и следовательно, фибриллин) выполняют
важную функцию. Они включают: эластичные стенки артерий (особенно аорты),
zonula
fibers глаза, связки, кожу и легочную паренхиму.
15. Опишите фенотип и
признаки поражения опорно-двигательного аппарата при синдроме Марфана.
БолЬным синдромом Марфана
присущ характерный фенотип. Их легко распознать благодаря высокому росту,
длинным тонким конечностям, арахнодактилии и сниженному количеству подкожной
жировой клетчатки.
Признаками поражения опорно-двигательного
аппарата являются: арахнодакти-лия ("пальцы паука"), воронкообразная деформация
грудной клетки, значительный рост, долихостеномелия (аномальное соотношение
верхнего и нижнего сегментов тела, < 0,85), сглаженность грудного кифоза,
сколиоз, готическое (высокое аркообразное) небо и долихоцефалия (узкое
удлиненное лицо)
Говорят, что несколько известных
атлетов и Авраам Линкольн имели синдром Марфана. Баскетбол и волейбол —
это те олимпийские виды спорта, в которых больные этим заболеванием достигают
больших результатов. Известно, что звезда американского волейбола, олимпийский
чемпион Фло Хайман умер в 1986 г. от сосудистых осложнений синдрома Марфана.
16. Как распознать арахнодактилию?
Посмотрите на руки больного.
Имеется три простых и достаточно точных признака для обнаружения арахнодактилии.
• Признак первого пальца,
или
симптом Штейнберга (Steinberg),— первый па-•I < лец выступает из-за
гипотенора при сжатом кулаке.
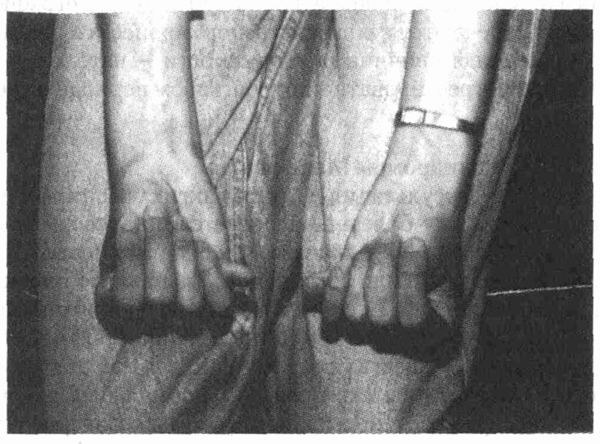
Пациент с синдромом Марфана демонстрирует
симптом Штейнберга (Из. The Clinical Slide Collection on the Rheumatic
Diseases. Atlanta, American College of Reumatology, 1991, с разрешения
)
• Признак кисти, или
симптом Уолкера-Мердока (Walker-Murdoch),— захождение первого пальца за
мизинец при схватывании кисти в области лучезапяст-ного сустава другой
руки.
• Пястный индекс —
рентгенологический признак арахнодактилии — средняя длина пясти, деленная
на среднюю ширину отрезка от 2-й до 4-й пястной кости, в норме составляет
5,4-7,9, а при синдроме Марфана > 8,4.
17. Является ли арахнодактилия
патогномоничным признаком синдрома Марфана? Какова дифференциальная диагностика?
Арахнодактилия отмечается
в 90 % случаев синдрома Марфана, но не является патогномоничным признаком,
так как наблюдается и при других заболеваниях. Неастеническая конституция
при синдроме Марфана может быть проявлением forme fruste1: больной
имеет арахнодактилию и нормальный фенотип при наличии той же генной мутации,
что и другие члены семьи с более тяжелыми проявлениями заболевания. При
синдроме морфаноподобной гиперподвижности суставов наблюдаются те же, что
и при синдроме Марфана, поражения скелета, например арахнодактилия, характерные
для синдрома Элерса-Данлоса сверхэластичная кожа и повышенная амплитуда
движений в суставах. Врожденная контрактурная арахнодактилия — это другое
аутосомно-доминантное заболевание неизвестной этиологии, при котором отмечаются
высокий рост, арахнодактилия и контрактуры суставов. Для больных, страдающих
гомоцистинурией, также характерны высокий рост, арахнодактилия и аномалии
позвоночника.
18. Какие клинические
проявления синдрома Марфана, кроме поражения опорно-двигательного аппарата,
известны? Какие из них являются причиной заболевания и смерти?
Вывих хрусталика всречается
у 50 % пациентов. Часто отмечаются сердечно-сосудистые расстройства. Наиболее
распространенной причиной смерти таких больных является разрыв аневризматически
расширенной восходящей аорты. Средняя продолжительность жизни при синдроме
Марфана — 32 года. Пролапс митрального клапана с регургитацией или недостаточность
аортального клапана диагностируют в 60 % случаев при аускультации сердца
и в 80 % при эхокардиографии. Поражения дыхательной системы встречаются
в виде кистозного процесса в легких и спонтанного пневмоторакса. Первичная
причина заболеваемости — патология позвоночника. Сколиоз может быстро прогрессировать
в подростковом периоде и требует хирургической коррекции.
19. Как лечить пациента
с синдромом Марфана?
Назначается генетическая
консультация и профилактика возникновения кардиологической патологии. Поскольку
болезнь наследуется по аутосомно-доминантному типу, то поражения возникают
в 50 % случаев. Необходимо провести эхокардиогра-фию для выявления митрального
или аортального клапанного порока. У больных с митральным или трикуспидальным
пороком не исключено возникновение инфекционного эндокардита, поэтому необходимо
проведение профилактических мероприятий. Эхокардиографию осуществляют ежегодно
для наблюдения за динамикой расширения аорты, а когда ее диаметр превысит
нормальный на 50 % — каждые полгода. Пропранолол сдерживает дилатацию аорты.
Тяжелая физическая нагрузка противопоказана. При отсутствии дилатации аорты
беременность для женщины безопасна.
Forme fruste (фр.) — приостановленная
болезнь.
20. Опишите клиническую
картину эластической псевдоксантомы.
Эластическая псевдоксантома
— редкое аутосомно-рецессивное заболевание, при котором происходят дегенерация
и кальцификация эластических волокон. Молекулярный дефект не установлен,
но предполагается, что он локализован где-то в структуре эластических волокон.
Клинические проявления этой болезни вариабельны. Изредка аутосомно-доминантная
форма клинически обнаруживается в зрелом возрасте. Классическим признаком
являются ксантоматоидные папулы, которые расположены на сгибательных
поверхностях конечностей. На глазном дне имеются ангиоид-ные полосы. Возможна
потеря зрения из-за макулопатии и поражения сетчатки. Не исключены кальцифицированные
депозиты в легких и поражения сердца, напоминающие кардиомиопатию. Отмечают
разрыв артерий желудочно-кишечного тракта. Наличие аномальных эластических
волокон не влияет на процесс заживления ран до достижения пациентом пожилого
возраста.
21. Что такое cutis
laxa (дряблая кожа)?
Cutis laxa — это группа
заболеваний, характеризующихся наличием дряблой, складчатой кожи. Кожа
кажется постаревшей, морщинистой со свободно свисающими складками. Подобная
картина иногда наблюдается после лечения воспалительных заболеваний кожи
пеницилламином или его производными. Кожа теряет эластичность, и кожная
складка не расправляется, в противоположность синдрому Элерса-Данлоса,
при котором кожная складка расправляется хорошо. Возможны поражения сердца,
легких — бронхоэктазы и эмфизема. Прочность кожи не изменяется (в отличие
от синдрома Элерса-Данлоса), что позволяет безопасно проводить хирургические
манипуляции.
22. Что такое синдром
Стиклера?
Синдром Стиклера (Stickler),
или наследственная артро-офтальмопатия,— наследственное заболевание неизвестной
этиологии, для которого характерны аномалии скелета и глазных яблок. Считается,
что это заболевание достаточно распространено и имеет аутосомно-доминантный
тип наследования. У 50 % больных (по данным одного исследования) имели
место аномалии коллагена II типа, коллагена хряща. Синдром включает миопию,
отслойку сетчатки и другие поражения глаз, расщепление твердого нёба, гипоплазию
нижней челюсти, повышенную и пониженную подвижность в суставах, дисплазию
эпифизов длинных трубчатых костей и развитие нетрудоспособности из-за поражений
суставов. Наличие данного заболевания следует предполагать у каждого больного
молодого возраста с дегенеративным артритом тазобедренного сустава или
у любого младенца с врожденно увеличенными в объеме суставами. Другими
клиническими синдромами, обусловленными аномалиями коллагена II типа, являются
первичный генерализованный остеоартрит, хондродиспла-зия, спондилоэпифизальная
дисплазия.
23. Какой молекулярный
дефект обнаруживают при синдроме Альпорта?
Синдром Альпорта включает
наследственный гломерулонефрит и глухоту. Дефектным является нефибриллярный
коллаген базальных мембран, коллаген IV типа. Коллаген VII типа, еще одна
структурная единица базальных мембран, участвует в процессе соединения
эпидермиса и дермы. При некоторых кожных заболеваниях, сопровождающихся
образованием пузырей, отмечают его аномалии.
Избранная литература
Barker D F Hostikka S L ,
Zhou J et al Identification of mutations m the COL4A5 gene in Alport syndrome
Science, 248 1224-1227,1990
Beighton P The dominant and
recessive forms of cutis laxa J Med Genet, 9 216-221,1972
Hermann J, France Т D, Spranger
J W et al The Stickler syndrome (hereditary arthroophthalmopathy) BirthDefects.il
76-103,1975
Komberg M , Auhcmo P Hand
and wrist joint problems in patients with Ehlers-Danlos syndrome J Hand
Surg , 10 193-196, 1985
Kuivamemi H, Tromp G, Prockop
D J Mutations in collagens Causes of rare and some common disorders in
humans FASEBJ.5 2052-2060,1991
Leier С V , Call Т D , Fulkerson
P К , Woolery С F The spectrum of cardiac defects in the Ehlers-Danlos
syndrome, types I and III Ann Intern Med , 92 171-178,1980
Marchase P, Holbrook R, Pmnell
S R A familial cutis laxa syndrome with ultra-structural abnormalities
of collagen and elastm J Invest Dermatol, 75 399-403,1980
Pope F M , Narcisi P , Nicholls
А С et al Clinical presentations of Ehlers-Danlos syndrome type IV Arch
Dis Child , 63 1016-1025,1989
Powell J Т, Adamson J, MacSweeney
S Т R et al Genetic variants of collagen III and abdominal aortic aneurysm
Eur J Vase Surg , 5 145-148,1991
Prockop D J Osteogenesis
imperfecta Model for genetic causes of osteoporosis and perhaps several
other common diseases of connective tissue Arthritis Rheum , 31 1-8, 1988
Pyentz R E Heritable disorders
of connective tissue In Schumacher H R (ed ) Primer on the Rheumatic-Diseases,
lOthed Atlanta, Arthritis Foundation, 1993 249-255
Pyentz R E Maternal and fetal
complications of pregnancy m the Marfan syndrome Am J Med, 71 784-790,1981
Pyentz R E , McKusick V A
The Marfan syndrome Diagnosis and management N Engl J Med, 300 772,1978
Rowe D W , Shapiro J R Disorders
of bone and structural proteins In Kelley W К , Harris E D , Ruddy S ,
Sledge С В (eds) Textbook of Rheumatology, 4th ed Philadelphia, W В Saunders,
1993,1567-1592
ГЛАВА 60. ВРОЖДЕННЫЕ
ДЕФЕКТЫ МЕТАБОЛИЗМА СОЕДИНИТЕЛЬНОЙ ТКАНИ
John Keith Jenkins, M.D.
i. Что такое гомоцистинурия?
Различают три клинические
(и биохимические) формы нарушения метаболизма аминокислот, которые характеризуются
повышением концентрации гомоцистина в плазме крови и моче Наиболее часто
встречающаяся форма гомоцистинурии связана с аутосомно-рецессивным дефектом
обмена метионина — отсутствием фермента цистатионин-р-синтазы, который
участвует в переносе сульфатного остатка от ме-тионина цистеину Две другие
более редкие формы — нарушение реметилирования гомоцистина до метионина
2. Часто ли встречается
гомоцистинурия?
Дефицит цистатионин-р-синтазы
в США встречается примерно 1 на 200 000 (или менее) младенцев, родившихся
живыми В некоторых штатах проводится скрининг на гомоцистинурию у всех
новорожденных Для сравнения, скрининг новорожденных на фенилкетонурию,
проводимый в большинстве штатов, выявляет 1 случай из 10 000 родившихся
живыми Гетерозиготы по дефициту цистатионин-р-синтазы (1 к 70 в общей популяции)
считаются здоровыми, однако могут входить в группу риска по раннему развитию
болезней периферической и церебральной сосудистых систем
3. Как диагностировать
гомоцистинурию?
При всех формах болезни отмечается
повышение концентрации гомоцистина в крови и моче Тест с цианидом-нитропруссидом
выявляет все аминокислоты, содержащие сульфгидрильную группу, однако этот
тест не специфичен при диагностике дефицита конкретного фермента Путь превращений
проходит от гомоцистина через метионин к цистеину Концентрация сывороточного
метионина повышена при дефиците цистатионин-р-синтазы, но нормальна или
снижена при нарушении обмена до стадии образования метионина Эти нарушения
также сопровождаются гомоцисти-нурией Другие патологические изменения метаболизма
обнаружены при синтезе кофакторов (метилвитамин В12 и метилтетрагидрофолат),
используемых в процессе образования метионина Необходимо измерять соотношение
концентраций гомоцистина и метионина в крови и моче, а затем проводить
точное определение тканевых ферментов (например из клеточной культуры фибробластов
кожи)
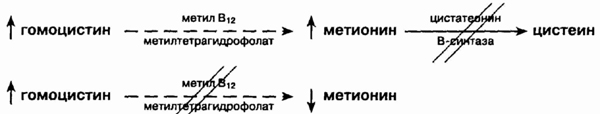
Биохимические пути, ведущие к гомоцистинурии
Вверху
Дефицит
цистатионин-р-синтазы ведет к повышению концентрации и гомоцистина и метионина
Внизу
Дефицит
других кофакторов ведет к повышению концентрации гомоцистина и снижению
концентрации метионина
4. Перечислите основные
клинические проявления гомоцистинурии.
• Вывих хрусталика (дислокация
хрусталика — патогномоничный признак)
• Тромбоэмболия
• Задержка умственного развития
• Заболевания соединительной
ткани
5. Какие нарушения опорно-двигательного
аппарата отмечают при гомоцистинурии?
Внешний вид больного напоминает
таковой при синдроме Марфана — высокий рост, арахнодактилия (долихостеномелия),
вывих хрусталика, деформации грудной клетки и позвоночника Встречается
также генерализованный остеопороз и тугоподвиж-ность суставов У больных
старше 15 лет остеопороз позвонков отмечается в 36-64 % случаев. Остеопороз
позвоночника выражен и сочетается с патологическими переломами, дегенеративными
изменениями дисков и межпозвонковых сочленений, сколиозом. Внутрисуставная
жидкость нормальная.
6. Какие поражения соединительной
ткани наблюдаются при гомоцистеинурии?
Цистеин необходим для правильного
поперечного соединения структурных белков (таких как коллаген и фибриллин)
соединительной ткани, костей, супрасенсорных связок глазного яблока, экстрацеллюлярного
матрикса эндотелиальных клеток. В связи с этим измененный коллаген может
стать причиной смещения хрусталика и остеопороза, в то время как аномальные
белки эластомера или его субструктур (фибриллин) лежат в основе фенотипического
сходства с синдромом Марфана. Измененное межуточное вещество эндотелиальных
клеток является причиной тромбозов и последующей задержки интеллектуального
развития.
7. Как лечить гомоцистинурию?
Какие симптомы будут исчезать при лечении?
Эффективное лечение требует
ранней диагностики заболевания с целью предупреждения задержки умственного
развития. Проведение скрининг-теста новорожденных — основа диагностики
врожденных нарушений метаболизма. Витамины В6, В12 и
фолаты являются кофакторами различных ферментов, участвующих в обмене ме-тионина.
Половина пациентов хорошо реагируют на терапию витамином В6,
высокие дозы которого снижают концентрацию метионина, увеличивают — цистеина
и улучшают состояние больных с дефицитом цистатионин-р-синтазы за счет
повышения активности небольшого количества резидуальных ферментов.
8. Регрессирует ли симптоматика
при терапии?
Прогрессирование эктопии
хрусталика, остеопороза и отставание умственного развития могут задерживаться
при лечении, но не исчезают, если процесс уже начался.
9. Каковы причины алкаптонурии?
Как наследуется это заболевание?
Алкаптонурия (или охроноз)
— это редко встречающееся нарушение метаболизма тирозина из-за дефицита
оксидазы гомогентизиновой кислоты. Этот фермент регулирует реакцию превращения
данной кислоты в форму, участвующую в цикле три-карбоновых кислот. Накопление
кислоты в тканях обусловливает их пигментацию от серой до темно-синей,
что и отражает название "охроноз". Вопрос об алкаптонурии — любимый на
экзаменах, так как это было первое описанное строго аутосомно-рецессивное
заболевание у человека с доказанным типом наследования. У гетерозигот симптоматика
не возникает даже при проведении нагрузочных проб предшественниками аминокислот.
10. Как диагностировать
алкаптонурию?
Диагностику облегчает характерная
триада симптомов:
• дегенеративный артрит (ранний);
• аномальная пигментация;
• моча при отстаивании изменяет
свой цвет на темно-синий (при ощелачивании
или добавлении хлорида железа).
Гомогентизиновая кислота
связывает коллаген и поэтому накапливается в соединительной ткани. Если
вы увидите у пациента темно-синие склеры, кожу, ушные раковины, хрящи,
подумайте об охронозе. Для лабораторного подтверждения диагноза проводится
специфический ферментный тест на оксидазу гомогентизиновой кислоты, а также
хроматография в тонком слое.
11. Опишите симптомы поражения
опорно-двигательного аппарата при алкаптон-урии.
Дегенеративный артрит проявляется
на третьем десятилетии жизни болью, тугопо-движностью и снижением амплитуды
движений в крупных суставах и позвоночнике. Наиболее часто поражаются позвоночник,
коленные суставы, тазобедренные и плечевые. Отмечаются их аномальная калыдификация
и оссификация, а также тенди-нит. Патогномоничной для заболевания считается
плотная кальцификация межпозвонковых дисков. Выявляют также симптом пустоты
в дисках. Синовиальная жидкость, в отличие от мочи, не изменяет своего
цвета, но имеет характерный перце-образный осадок. Замечено, что болезнь
накопления пирофосфата кальция может быть сопутствующим заболеванием.
12. Как лечить охронозный
артрит?
Стандартная терапия — это
симптоматическое лечение, такое же, как при остеоарт-рите: аналгезия, физиотерапия,
местные воздействия и т. д. Показан прием пищи с низким содержанием фенилаланина
и тирозина. Хирургические манипуляции (включая артройластику и артроскопию)
с успехом использовали для удаления свободно лежащих в полости сустава
костно-хрящевых тел. Проводилось лечение витамином С в высоких дозах, но
исследований, подтверждающих его эффективность, нет.
13. Что такое синдром
курчавых волос Менкеса? Как заболевание наследуется?
Синдром курчавых волос Менкеса
(Menkes) — это сцепленное с Х-хромосомой рецессивное нарушение обмена меди,
клинически выражающееся неврологическими проявлениями: судорогами, аномальными
рефлексами, спастичностью мышц, задержкой умственного развития и скрученными
(четкообразными, хрупкими и редкими) волосами.
14. Какова причина поражений
опорно-двигательного аппарата при данном синдроме?
Измененный метаболизм меди
вызывает нарушение функции медь-зависимых ме-таллоферментов, участвующих
в синтезе коллагена и эластина, что приводит к поражению соединительной
ткани. Повреждения опорно-двигательного аппарата могут напоминать таковые
либо при синдроме cutis laxa, либо при синдроме Элерса-Дан-лоса,
то есть "сверхрастяжимости" кожи и суставов.
Избранная литература
Albers S. Е., Brozena S.
J., Glass R., Fenske N. A. Alkaptonuna and ochronosis- Case report and
review. J. Am. Acad. Dermatol., 27: 609-614, 1992
Gordan D. A. Storage and
deposition diseases In- Schumacher H R. (ed) Primer on the Rheumatic Diseases,
10th ed. Atlanta, Arthritis Foundation, 1993, 227-228
Hunter Т., Gordon D , Ogryzlo
M. A The ground pepper sign of synovia! fluid A new diagnositic feature
of ochronosis J. Rheumatol., 1 45-53, 1974
Mudd S. H., Skovby F., Levy
H. L. et al. The natural history of homocystmuna due to cystathionme beta-synthase
deficiency Am. J. Hum Genet., 37:1-31, 1985.
Pyeritz R. E. Heritable disorders
of connective tissue. In: Schumacher H. R. (ed.) Primer on the Rheumatic
Diseases, 10th ed. Atlanta, Arthritis Foundation, 1993, 249-255
Rowe D W., Shapiro J. R Disorders
of bone and structural proteins. In: Kelley W. K., Harris E D., Ruddy S.,
Sledge С В (eds). Textbook of Rheumatology, 4th ed. Philadelphia, W В Saunders,
1993,1567-1592.
Sakkas L .Thomas В., Smyrms
P., Vlahos E. Low back pain and ochronosis. Int. Orthop., 11:19-21,1987
ГЛАВА 61. БОЛЕЗНИ
НАКОПЛЕНИЯ
MarkJarek, M.D., FACR
1. Каков тип наследования
первичного гемохроматоза? Болезни Вильсона? Ал-каптонурии? *
Эти заболевания наследуются
по аутосомно-рецессивному типу. Гетерозиготы являются бессимптомными носителями
дефектного гена. Болезнь Вильсона встречается приблизительно в таком соотношении
1:30 000, а алкаптонурия — 1: 200 000. Алкап-тонурия была первым заболеванием,
для которого доказан аутосомно-рецессивный тип наследования.
2. Какие антигены лейкоцитов
человека (HLA) связаны с гемохроматозом?
Генетическая основа гемохроматоза
открыта Симоном (Simon) и соавт. в 1976 г. Ген гемохроматоза находится
на коротком плече шестой хромосомы около HLA-A-локу-са. HLA-АЗ-аллоантиген
обнаруживается у 70 % пациентов с гемохроматозом и приблизительно у 20
% лиц белой расы (относительный риск равен 23). Основа этой связи кроется
в мутации, произошедшей у древних народов северной Европы, вероятно, из-за
сниженного поступления в организм железа. HLA-A2, А29, В7 и В14 — другие
гаштотипы, связанные с гемохроматозом.
3. Как часто встречается
гемохроматоз?
Скрининговые исследования
позволили установить, что ген гемохроматоза встречается у 5 % лиц белой
расы, частота носительства гена (гетерозиготы) достигает приблизительно
10 %, а заболеваемость (гомозиготы) — 1-3:1000. Исследования в скандинавских
семьях показали, что гомозиготами является почти 1 % населения. Таким образом,
гемохроматоз — одно из самых частых наследственных метаболических заболеваний.
Клиническое проявление зависит от нескольких факторов. Наиболее часто это
— кровотечение у женщин, связанное с менструациями и беременностью. (Увеличенный
объем кровотечения при травме — в пять раз более частый симптом заболевания
у мужчин, чем у женщин.) Данная частота гемохроматоза в общей популяции
заставляет врачей проводить скрининг-исследование на содержание железа
у всех мужчин старше 40 лет.
4. Как выглядит типичный
больной гемохроматозом?
Признаки заболевания начинают
проявляться в возрасте 40-60 лет, при этом тяжесть его вариабельна. У некоторых
больных можно обнаружить все клинические проявления болезни, в то время
как у других гомозигот они никогда не развивались.
Обычно гемохроматоз проявляется
патологическими
показателями анализов функции печени или гепатомегалией.
Без лечения
поражение печени часто приводит к циррозу. Характерная для заболевания
артропатия встречается в 20-50 % случаев и может быть первым ее проявлением,
но чаще артропатия развивается позже и возникает уже после начала лечения.
Другими признаками болезни являются темно-серая или коричневая
пигментация
кожи (накопление меланина), сахарный диабет и гипогонадизм:
снижение
либидо, импотенция, редкий волосяной покров тела, аменорея. Часто отмечаются
системные
симптомы, такие как мышечная слабость, заторможенность, вялость. Поражение
сердца, как правило, проявляется застойной сердечной недостаточностью
(у
30 % пациентов) и становится непосредственной причиной смерти при отсутствии
лечения.
5. Опишите артропатию
при гемохроматозе.
Поражаются многие суставы,
но характерны жалобы на боли и тугоподвижность в пястно-фаланговых суставах
второго и третьего пальцев. Не исключено вовлечение в процесс проксимальных
межфаланговых суставов, лучезапястного, коленного, тазобедренного, голеностопного,
плечевого и, изредка, плюснофаланговых. При осмотре сустава отмечают твердый
отек с небольшой болезненностью при пальпации, но гиперемия и повышение
температуры сустава не определяются, что и помогает отличить артроз при
гемохроматозе от ревматоидного артрита.
6. Опишите характерную
рентгенологическую картину артроза при гемохроматозе.
На рентгенограмме выявляют
остеоартритоподобные изменения: склеротический процесс по краям костей,
сужение суставной щели и образование характерных крюч-ковидных остеофитов,
особенно в пястно-фаланговых суставах. Хондрокальциноз встречается у 30-60
%
больных
без дегенеративной артропатии.
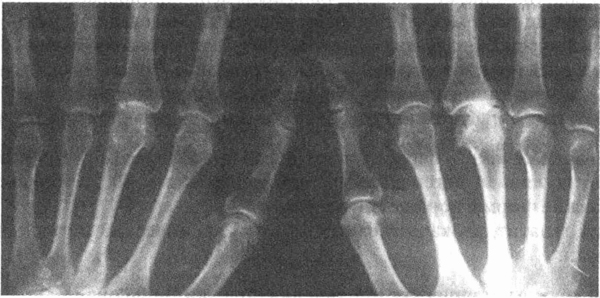
Рентгенограмма кисти больного гемохроматозом.
Отмечается дегенеративный артрит пястно-фаланговых суставов с крючковидными
остеофитами
7. Распространенная остеопения
часто встречается при гемохроматозе. Каковы ее причины?
• Повышенное содержание железа
в синовиальной жидкости (по сравнению с сывороткой) тормозит формирование
кости.
• Инфильтрация железом гипофиза
снижает секрецию гонадотропина, что приводит к гипогонадизму.
• Цирроз печени спровождается
атрофией яичка и гипогонадизмом.
8. Каков общий план лечения
гемохроматоза?
Кровопускание проводится
один-два раза в неделю до достижения нормальной концентрации трансферрина
и ферритина (2-3 года), а затем эта процедура выполняется периодически
в зависимости от содержания. Продолжительность жизни пациентов с симптомами
заболевания увеличивается значительно после удаления чрезмерных накоплений
иона железа (5-летняя выживаемость составляет 90 % и без лечения — 33 %).
При лечении уменьшается гепатомегалия, анализы функции печени приходят
к норме, уменьшается пигментация, сердечная недостаточность стабилизируется
или ее выраженность уменьшается. Проявления сахарного диабета уменьшаются
в 50 % случаев. Кровопускание не влияет на развитие гипогонадизма или артропатии.
Фиброз печени может уменьшаться, но цирроз не претерпевает обратного развития.
Вероятность возникновения печеночно-клеточной карциномы, которая относится
к поздним осложнениям у пациентов с циррозом печени, не уменьшается. Продолжительность
жизни гомозигот, у которых был диагностирован гемохроматоз и начато его
лечение до развития цирроза печени, такая же, как в нормальной популяции,
поэтому значение семейного скрининга и раннего начала лечения трудно переоценить.
Применение хелатов и антиоксидантов типа а-токоферола дает некоторый положительный
эффект у пациентов с поражением сердца.
9. Кому из родственников
следует провести скрининговое исследование, если пробанду поставлен диагноз
гемохроматоза?
Всем родственникам первого
уровня (старше 10 лет) следует проводить скрининг каждые 2-5 лет, измерять
насыщение трансферрина (содержание железа/общая железо-связывающая емкость
[ОЖСЕ]). Если этот показатель превышает 50 %, определяют ферритин в сыворотке
крови. При повышенном содержании ферритина проводят биопсию печени с определением
в ткани количества железа. Из-за высокой генетической связи между родственниками
первого уровня проведение типирования по HLA-системе в качестве скрининга
может стать дорогостоящим исследованием, учитывая индекс "цена-эффективность".
Болеющие сибсы (братья и сестры) обычно имеют оба HLA-ran-лотипа, идентичные
тем, которые выявляются у пробанда. Сибсы только с одним гапло-типом HLA
обычно не имеют клинических признаков накопления железа. Если заболевают
дети пробанда, то, возможно, произошло гомозиготно-гетерозиготное скрещивание.
10. Заболевание печени
— первое клиническое проявление болезни Вильсона (ге-патолентикулярная
дегенерация) приблизительно у 50 % больных. Каковы клинические формы поражения
печени?
Преходящий гепатит, злокачественный
гепатит, хронический активный гепатит и цирроз печени.
11. Каковы другие клинические
проявления болезни Вильсона?
В дополнение к симптомам
поражения печени при болезни Вильсона, которая обычно проявляется в возрасте
от 20 до 40 лет, обнаруживают неврологические расстройства, артропатию
и гемолитическую анемию. Частыми неврологическими расстройствами являются
нарушения движений вследствие дегенерации ядер двигательных нервов, но
описаны и другие синдромы, в том числе психиатрические. К гинекологическим
проявлениям заболевания относятся аменорея и бесплодие.
12. Опишите кольцо Кайзера-Флейшера.
Каково его значение?
Кольцо Кайзера-Флейшера (Kayser-Fleischer)
— это депозиты меди зеленого или коричневого цвета в мембране Децемета,
не влияющие на зрение. Его наличие — точное предсказание неврологических
и психиатрических расстройств, поэтому если оно при исследовании на щелевой
лампе у больного с явными неврологическими и психиатрическими синдромами
отсутствует, то это не болезнь Вильсона. Кольцо Кайзера-Флейшера изредка
наблюдается и при других заболеваниях, сопровождающихся циррозом печени,
поэтому не специфично для болезни Вильсона.
13. Какое биохимическое
нарушение лежит в основе болезни Вильсона? Как поставить диагноз?
Болезнь Вильсона развивается
из-за избыточного накопления меди, сочетающегося с дефицитом церулоплазмина.
Когда способность гепатоцитов накапливать медь исчерпывается, она начинает
откладываться в строме печени и в других органах, таких как мозг, почки;
увеличивается содержание меди в моче и сыворотке крови. Снижение концентрации
сывороточного церулоплазмина (< 20 мг/дл) и меди (< 80 мкг/дл) и
повышение почечной экскреции меди (> 100 мкг/сут) характерно для болезни
Вильсона. Повышение концентрации меди в печени (> 250 мкг/г ткани) — надежный
признак для ранней диагностики заболевания.
14. Офтальмолог обнаружил
у молодого человека, жалующегося на артрит, кольца Кайзера-Флейшера. Какие
еще признаки поражения опорно-двигательного аппарата можно выявить у данного
пациента?
Боль и отек пястно-фаланговых,
лучезапястных, локтевых, плечевых, коленных и тазобедренных суставов, напоминающие
поражения при гемохроматозе; не исключены такие же бессимптомные изменения
рентгенологической картины.
15. Какова рентгенологическая
картина при поражении сустава при болезни Вильсона?
Субхондральная концевая и
кортикальная фрагментация кости, субхондральный и центральный склероз костей
лучезапястного, локтевого, плечевого, коленного суставов и суставов кисти
позволяют отличить эту артропатию от остеоартрита. Поражение тазобедренных
и пястно-фаланговых суставов, в отличие от гемохроматоза, встречается редко.
Еще реже обнаруживают расслаивающий остеохондрит, хондро-кальциноз, хондромаляцию
надколенника, клиновидную деформацию позвонков и распространненый остеопороз
или остеомаляцию.
16. Остеопения и/или остеомаляция
обнаруживаются у 25-50 % пациентов с болезнью Вильсона. Каков возможный
механизм их возникновения?
Остеопения и остеомаляция
— возможно, результат почечного канальцевого ацидоза или синдрома Фанкони,
которые являются частыми поражениями почек при болезни Вильсона.
17. Как лечить пациента
с болезнью Вильсона?
Длительная терапия хелатами
(пеницилламином) действенно предупреждает любое проявление болезни. Если
в процессе лечения развивается необратимая непереносимость пеницилламина,
используют триентин или проводят пересадку печени.
18. Какой биохимический
дефект лежит в основе алкаптонурии?
Алкаптонурия — это нарушение
обмена тирозина в результате дефицита оксидазы го-могентизиновой кислоты
(2,5-(диоксифенил)-уксусная кислота — промежуточный продукт катаболизма
тирозина), приводящего к экскреции большого количества данной кислоты с
мочой и накоплению окисленной гомогентизиновой кислоты (пигмента) в соединительной
ткани (охроноэ). Характерной чертой болезни является потемнение мочи при
стоянии из-за большой концентрации в ней гомогентизиновой кислоты.
19. Перечислите пять мест,
где может накапливаться пигмент при охронозе.
Депозиты серо-коричневого
пигмента при охронозе обнаруживают в коже, склере, стенках артерий, предстательной
железе и ушах (ушная раковина, ушная сера, про-тивозавиток ушной раковины).
Это приводит к стенозу аорты, нарушению проведения звука и образованию
конкрементов в предстательной железе. Накопление пигмента в суставных хрящах
и межпозвонковых дисках в конечном счете выражается в развитии охронозной
артропатии.
20. Опишите клинические
черты охронозной артропатии.
Охроноз обычно проявляется
в возрасте от 20 до 30 лет как прогрессирующий спон-дилез со снижением
объема движений в пояснично-крестцовом отделе позвоночника. У больных в
положении стоя поза напоминает такую же при анкилозирующем спондилите —
сутулая спина, отсутствие поясничного лордоза, снижение роста, согнутые
нижние конечности в тазобедренных и коленных суставах, но при этом отсутствуют
характерные для анкилозирующего спондилита кольцевая оссификация и анкилоз
пояснично-крестцового соединения. Накопление пигмента при охронозе в пульпозном
ядре предрасполагает к образованию грыжи межпозвонкового диска, что может
проявляться как острое начало синдрома боли в нижней части спины, клинически
неотличимого от боли при грыже диска без алкаптонурии. Дегенеративный артрит
периферических суставов встречается реже, чем болезни позвоночника. Наиболее
часто поражаются коленные, плечевые и тазобедренные суставы. Так же как
и при артропатии, при гемохроматозе больные жалуются на боль, тугоподвижность
суставов, ограничение амплитуды движений. Однако при охронозе не поражаются
лучезапястный сустав, мелкие суставы кисти и стопы.
21. Какова характерная
рентгенологическая картина позвоночника при охронозе?
На рентгенограмме пояснично-крестцового
отдела позвоночника можно заметить ранние дегенеративные изменения: плотная
кальцификация межпозвонковых дис-
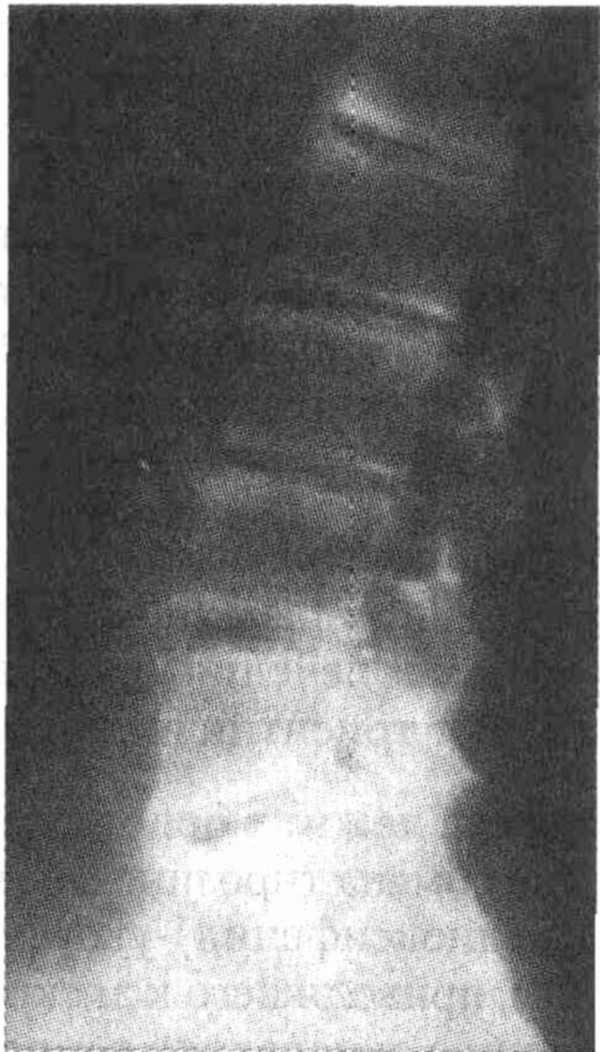
Боковая рентгенограмма позвоночника
больного охронозом Отмечается межпозвонковой щели на многих уровнях
ков и сужение межпозвонковых
щелей. Выраженная кальцификация межпозвонковых дисков обнаруживается также
при гемохроматозе, гиперпаратиреозе, болезни накопления пирофосфата кальция,
паралитическом полиомиелите и амилоидозе. Рентгенологическая картина поражения
крупных суставов при охронозном артрите практически такая же, как и при
первичном остеоартрите.
22. Существует ли эффективное
лечение охроноза?
Предложено много способов
лечения: введение аскорбиновой кислоты, назначение диеты с пониженным содержанием
белка, фенилаланина и тирозина, но на данный момент терапии с отчетливым
положительным эффектом нет.
Избранная литература
Bothwell Т. Н., Charlton
R. W., Cook J. D., Finch С. A. Iron Metabolism in Man. Oxford, Blackwell,
1979.
Bothwell T. H, Charlton R.
W, Motulsky A. G. Hemochromatosis. In: Scnver С. Н., Beau-det A. L, Sly
W. S. et al. (eds). The Metabolic Basis of Inherited Disease, 6th ed. New
York, McGraw-Hill, 1989, 1433-1462.
Edwards C. Q., Kushner J.
P. Screening for hemochromatosis. N. Engl. J. Med., 328:1616-1620, 1993
Edwards C. Q., Skolnich
M. H., Kushner J. P. Hereditary hemochromatosis: Contributions of genetic
analysis. Prog. Hematol., 12:43-71,1981.
Gordon D. A. Storage and
deposition diseases. In: Schumacher H. R. (ed.). Primer on the Rheumatic
Diseases, 10th ed. Atlanta, Arthritis Foundation, 1993, 225-230.
Lalouel J. M., Jorde L. B.
Idiopathic hemochromatosis: Significance and implications of linkage and
association to HLA. Ann. N. Y. Acad. Sci., 526: 34-46, 1988.
Lambert R. E., McGuire J.
L. Iron storage disease. In: Kelly W. N.. Hams E. D., Ruddy S., Sledge
С. В. (eds). Textbook of Rheumatology, 4th ed. Philadelphia, W B. Saunders,
1993,1435-1443.
Niederau C., Fischer R.,
Sonnenberg A., Stremmel W et al. Survival and causes of death in cirrhotic
and noncirrhotic patients with primary hemochromatosis. N. Engl. J. Med.,
313: 1256-1262,1985.
Olsson K. S. Hemochromatosis.
In: Khppel J. H., Dieppe P. A. (eds). Rheumatology, London, Mosby, 1994,7:18.1-7:18.4.
Powell L. W., Isselbacher
K. J. Hemochromatosis. In: Wilson J. D. et al. (eds). Harnson's Principles
of Internal Medicine, 12th ed. New York, McGraw-Hill, 1991,1825-1829.
Resnick D., Berthiaume M.
J., Sartons D. Diagnostic tests and procedures m rheumatic diseases. In.
Kelly W. N., Harris E. D., Ruddy S., Sledge С В (eds). Textbook of Rheumatology,
4th ed , Philadelphia, W В Saunders, 1993, 627-628.
Rosenberg L. E Storage diseases
of ammo acid metabolism. In. Wilson J. D. et al. (eds). Harnson's Principles
of Internal Medicine, 12th ed., New York, McGraw-Hill, 1991, 1875-1876.
Scheinberg I. H Wilson's
disease. In: Wilson J D. et al. (eds). Harnson's Principles of Internal
Medicine, 12th ed., New York, McGraw-Hill, 1991, 1843-1845.
Schumacher H R Ochronosis,
hemochromatosis, and Wilson's disease. In' McCarty D. J., Koopman W. J.
(eds). Arthritis and Allied Conditions, 12th ed., Philadelphia, Lea &
Febiger, 1993,1913-1925
' Baer D. M., Simons J. L.,
Staples R. L. et al. Hemochromatosis screening m asymptomatic "' ambulatory
men 30 years of age and older Am. J. Med., 98:464-468, 1995
ГЛАВА 62. РЕВМАТИЧЕСКИЕ
ПРОЯВЛЕНИЯ СИНДРОМОВ ПЕРВИЧНОГО ИММУНОДЕФИЦИТА
Mark Malyak,M.D.
1. Почему ревматологу
необходимо знание синдромов первичного иммунодефицита?
Синдромы первичного иммунодефицита
могут быть связаны с различными проблемами в дополнение к повышенному риску
возникновения инфекций, включая аутоиммунные состояния, аллергию, возможность
развития лимфоидных и эпителиальных опухолей. Аутоиммунные процессы могут
проявляться определенными аутоиммунными заболеваниями, например системной
красной волчанкой при врожденном дефиците С4. С другой стороны, различные
антитела присутствуют, но нет клинических проявлений аутоиммунной болезни
(ревматоидный фактор или анти-нуклеарные антитела при избирательном дефиците
IgA).
2. Какие составные части
иммунной системы поражаются при синдромах первичного иммунодефицита?
• В-клетки (гуморальный иммунодефицит).
• Т-клетки (клеточный иммунодефицит).
• Клетки-киллеры.
• Фагоциты.
• Белки комплемента.
Синдром первичного иммунодефицита
может возникать или из-за дисфункции одного компонента иммунной системы,
как при дефиците С4, или вследствие нарушений нескольких составляющих —
поражение В-клеток, Т-клеток и функции фагоцитов при определенных тяжелых
комбинированных иммунодефицитах.
3. Определение типа рецидивирующей
инфекции помогает в диагностике причинного синдрома иммунодефицита. Какие
микроорганизмы ответственны за развитие рецидивирующего инфекционного процесса
при синдромах В-клеточно-го иммунодефицита?
В-клеточный иммунодефицит,
например сцепленная с Х-хромосомой агаммаглобу-линемия (Брутона [Bruton]),
выражается в неадекватной продукции иммуноглобу-линов, что сопровождается
возникновением рецидивирующего инфекционного процесса, возбудителями которого
являются внеклеточные, гноеродные бактерии, имеющие оболочки, особенно
Streptococcus
pneumoniae и Haemophilus influenzae. Эти микроорганизмы, как
правило, вызывают острые и хронические инфекции верхних и нижних отделов
дыхательных путей, менингит и сепсис.
4. Какие организмы вызывают
инфекционный процесс при Т-клеточном иммунодефиците, таком как гипоплазия
вилочковой железы (синдром Ди Георге [Di George])?
• Вирусы (герпес-вирусы).
• Внутриклеточные бактерии
(микобактерии).
• Грибки (виды Candida,
Pneumocystis carinii).
Первичный Т-клеточный иммунодефицит
проявляется нарушением Т-клеточного иммунитета, что ведет к возникновению
инфекций, подобных тем, что встречаются
у больных с ВИЧ-инфекцией,—
прототипом приобретенного Т-клеточного иммуно-дефицитного состояния.
5. Назовите лабораторные
тесты для определения состояния гуморальной иммунной системы (функции В-клеток).
Лабораторное определение функции
В-клеток
|
|
|
|
|
|
КАТЕГОРИЯ
|
СПЕЦИФИЧЕСКИЕ
ТЕСТЫ
|
КОММЕНТАРИИ
|
|
|
Функциональные
тесты in vivo
|
Титры изогемагглютининов
(против А- и В-группы)
|
Обычно встречающиеся;
преимущественно IgM
|
|
|
(стандартные
скрининговые тесты)
|
Иммунизация
против дифтерии и столбняка
|
Определение
антител до и через 2 нед; исследуется способность синтезировать IgG против
белковых антигенов
|
|
|
|
Иммунизация
против пневмококка
|
Определение
антител до и через 3 нед; исследуется способность синтезировать антитела
против полисахаридных антигенов
|
|
|
Определение
|
Содержание
IgM,
IgG, IgA
|
Используют
различные иммунные тесты
|
|
|
количества
|
Содержание
подклассов IgG,
|
ФИС, РИТ
|
|
|
иммуиоглобулинов
|
концентрация
IgE
|
|
|
|
Тесты in
vitro (дорогостоящие)
|
Подсчет В-клеток
в периферической крови
|
Могут использоваться
анти-1д-антитела и специфические моноклональные антитела
|
|
|
|
Подсчет В-клеток
в пунктате костного мозга
|
Измеряют
поверхностные 1д-негативные, цитоплазматические т-цепочечно-пози-тивные
клетки
|
|
|
|
Синтез иммуноглобулинов
in
vitro
|
Мононуклеарные
клетки периферической крови, стимулированные митогеном фито-лаки американской
(Phutolacca
americana)
|
|
|
|
|
|
|
ФИС — ферментная иммуносорбция; РИТ
— радиоиммунные тесты.
Целесообразно для диагностики
функции В-клеток определить уровень IgA в сыворотке крови и провести недорогостоящие
функциональные тесты in vivo. Если результаты тестов нормальные,
клинически значимое нарушение функции В-клеток можно исключить. Если результаты
тестов патологические, проводят количественное определение IgG и IgM и,
по возможности, тесты in vitro. Эти исследования необходимы для
выявления причины лежащего в основе нарушения синдрома первичного иммунодефицита.
6. Какими лабораторными
тестами определяют состояние клеточного иммунодефицита (функции Т-клеток)?
Для диагностики функции Т-клеток
целесообразно определить абсолютное число лимфоцитов и провести кожный
тест с Candida. Если результаты нормальны, клинически значимое нарушение
функции Т-клеток может быть исключено. Если кожный тест с Candida отрицательный,
то нужно провести исследование с остальными четырьмя антигенами для определения
нарушения Т-клеточного иммунитета. Если результаты патологические, понадобятся
сложные тесты in vitro для определения базисного нарушения Т-клеточного
иммунитета. Анализ на ВИЧ-инфекцию проводят как часть скринингового исследования
для исключения этого приобретенного Т-клеточного заболевания.
Лабораторное определение функции
Т-клеток
|
|
|
|
|
|
КАТЕГОРИЯ
|
СПЕЦИФИЧЕСКИЕ
ТЕСТЫ
|
КОММЕНТАРИИ
|
|
|
Функциональные
тесты in vivo (кожные тесты для определения ОГ) (стандартные скрининговые
тесты)
|
Кожный тест
на Candida Реакция Манту, на Tnchophy-ton, свинку, столбнячный/
дифтерийный токсин
|
Определяется
уровень индуративного отека спустя 24-72 ч Если тест на Candida отрицательный,
необходимо проведение теста по крайней мере с 4 антигенами для диагностики
состояния клеточного иммунитета
|
|
|
Определение
абсолютного числа лимфоцитов (стандартные скрининговые тесты)
|
Определение
общего числа лейкоцитов и количества лимфоцитов
|
Тяжелое нарушение
клеточного иммунитета маловероятно, если число лимфоцитов нормальное
|
|
|
Тесты /л
vitro
(дорогостоящие)
|
Определение
числа: всех Т-клеток клеток CD4* клеток CD8* клеток-киллеров
|
Могут использоваться
специфические моноклинальные антитела
|
|
|
|
Реакция бласттрансформации
лимфоцитов
|
Оценка потребления
меченого тимидина после стимуляции клеток лектинами (такими как ФГА), специфическими
антигенами (такими как Candida) или однонаправленная реакция всех
лимфоцитов
|
|
|
|
Количественное
определение способности Т-клеток синтезировать интерлейкин-2 и интерлейкин-2-рецепторы
|
Этот тест
и реакция бласттрансформации лимфоцитов — индикаторы хорошей активации
Т-клеток
|
|
|
|
|
|
|
ФГА — фитогемагглютинин, ОГ — отсроченная
гиперчувствительность
7. Какие микроорганизмы
вызывают септический артрит у пациентов с гипогам-маглобулинемией вследствие
первичного В-клеточного иммунодефицита?
Избирательный дефицит IgA,
сцепленная с Х-хромосомой (Брутона) агаммаглобули-немия, простой вариабельный
иммунодефицит (ПВИ) и дефицит иммуноглобулинов с повышенным содержанием
IgM (гипер-IgM) составляют более 99 % всех первичных гипогаммаглобулинемических
состояний (В-клеточный иммунодефицит). Больные подвержены возникновению
септического артрита, возбудителями которого являются обычные микроорганизмы,
наблюдаемые при В-клеточных иммунодефицитах: Streptococcus рпеитотае,
Haemophilus influenzae и Staphylococcus aureus. Кроме этих типичных
бактерий, воспаление суставов вызывают Ureaplasma mealyticum и другие
микроорганизмы рода Mycoplasma. Частота возникновения септического
артрита при В-клеточном иммунодефиците неизвестна, но она меньше таковой
распространенных инфекций верхних и нижних дыхательных путей и желудочно-кишечного
тракта.
8. Какие из синдромов
первичного иммунодефицита наиболее часто связаны с аутоиммунными заболеваниями?
Избирательный дефицит IgA,
сцепленная с Х-хромосомой (Брутона) гаммаглобули-немия, простой вариабельный
иммунодефицит и дефицит иммуноглобулинов с повышенным содержанием IgM (гипер-IgM).
Полное отсутствие определенных компонентов комплемента (С2, С4) также характерно
для возникновения этих болезней, особенно системной красной волчанки Более
того, хроническая гранулематозная болезнь, первичная болезнь нейтрофилов,
определяется наличием антинуклеарных антител и, реже, связана с СКВ. Как
правило, у больных с преобладанием поражения Т-клеточного иммунитета не
отмечается аутоиммунных заболеваний, возможно из-за того, что эти больные
редко переживают период новорожденности.
9. Каковы ревматологические
проявления сцепленной с Х-хромосомой агамма-глобулинемии?
Ревматологические проявления сцепленной
с Х-хромосомой агаммаглобулинемии
|
|
|
|
|
Септический
артрит Внеклеточные бактерии (S pneumoniae, H. mfluenzae, S. aureus)
Mycoplasma, особенно Ureaplasma urealyticum Энтеровирусы, особенно
ЕСНО-вирусы и вирусы Коксаки
|
Встречается
в 20 % случаев
|
|
|
Асептический,
возможно аутоиммунный, артрит
|
Обычно поражает
один или два крупных сустава, деструктивный; отсутствуют ревматоидный фактор
и антинуклеар-ные антитела
|
|
|
Дерматомиозитоподобный
синдром, связанный с прогрессирующей энтеровирусной инфекцией ЦНС
|
Проявляется
сыпью и мышечной слабостью
|
|
|
|
|
|
Сцепленная с Х-хромосомой
(Брутона) агаммаглобулинемия — редкое заболевание, характеризующееся отсутствием
или очень малой концентрацией сывороточных IgG, IgM, IgA и патологическими
В-клеточными функциональными тестами in vivo Система клеточного
иммунитета интактна. Поражение клеток обусловлено нарушением созревания
В-клеточной линии, поэтому в периферической крови нет В-клеток. Артрит
встречается приблизительно у 20 % больных и в половине случаев вызван обычными
гноеродными бактериями. Кроме того, больные высокочувствительны к энтеровирусной
и микоплазменной инфекции.
Иногда при артрите на фоне
сцепленной с Х-хромосомой (Брутона) агаммаглобулинемии не удается обнаружить
инфекционного возбудителя, несмотря на тщательную диагностику. Эти случаи
могут быть обусловлены редкой инфекцией, которая не идентифицируется, или
представлять собой истинное аутоиммунное заболевание (ювенильный ревматоидный
артрит). В целом аутоиммунные процессы встречаются значительно реже при
сцепленной с Х-хромосомой (Брутона) агаммаглобулинемии, чем при избирательном
дефиците IgA или простом вариабельном иммунодефиците.
10. Каковы ревматологические
проявления избирательного дефицита IgA?
• Наличие аутоантител, особенно
ревматоидного фактора и антинуклеарных антител при отсутствии выраженной
симптоматики аутоиммунной болезни.
• Системные аутоиммунные
заболевания (СКВ, асептический, возможно аутоиммунный артрит и др.).
• Органоспецифичные иммунные
заболевания (сахарный диабет I типа, myas-
thenia gravis и др.).
Избирательный иммунодефицит
IgA — наиболее часто встречающийся синдром первичного иммунодефицита, распространенность
его в общей популяции составляет почти 1: 330. Он характеризуется отсутствием
или очень низким содержанием в сыворотке крови IgA при нормальном содержании
— IgG и IgM. Клеточный иммунитет не нарушен. Пациенты могут не предъявлять
серьезных жалоб, но болеть рецидивирующими инфекциями дыхательных путей
и желудочно-кишечного тракта или аутоиммунными заболеваниями. В большинстве
случаев избирательный иммунодефицит IgA, вероятно, обусловлен генетическим
дефектом, который проявляется при рождении и сохраняется в течение всей
жизни. Некоторые случаи носят характер приобретенного заболевания вследствие
лекарственной терапии или вирусной инфекции и обычно преходящи.
11. Какие аутоантитела
обнаруживают у больных избирательным иммунодефицитом IgA без клинически
выраженного заболевания?
Наличие аутоантител без клинически
выраженного заболевания встречается часто при избирательном иммунодефиците
IgA. Чаще всего обнаруживают ревматоидный фактор и антинуклеарные антитела,
реже — антитела к дву- и односпиралевой ДНК, кардиолипиновые, тироглобулиновые,
антитела к микросомам щитовидной железы, гладкомышечной ткани, париетальным
клеткам желудка, поперечно-полосатой мышечной ткани, ацетилхолиновым рецепторам,
желчным протокам. Аутоантитела против IgA встречаются почти у 44 % пациентов.
12. Какие из системных
и органо-специфичных аутоиммунных заболеваний связаны с синдромом избирательного
иммунодефицита IgA?
|
|
|
|
|
СИСТЕМНЫЕ
|
ОРГАНОСПЕЦИФИЧНЫЕ
|
|
|
Ситемная
красная волчанка' Ювенильный ревматоидный артрит1 Ревматоидный
артрит' Синдром Шегрена Склеродермия Дерматомиозит Синдромы васкулита
|
Сахарный
диабет 1 типа' Myasthenia gravis'1 Воспалительные заболевания
кишечника Аутоиммунный гепатит Пернициозная анемия Первичная недостаточность
надпочечников
|
|
|
|
|
|
'Наиболее вероятная связь. Описаны
и другие заболевания, но наличие связи с дефицитом IgA требует доказательства.
13. Каковы ревматологические
проявления простого вариабельного иммунодефицита?
Ревматологические проявления простого
вариабельного иммунодефицита
Септический артрит
Внеклеточные бактерии с клеточной стенкой
(S. pneumoniae, H. influenzae, S. aureus)
Mycoplasma (особенно Ureaplasma
urealyticum) Асептический, возможно аутоиммунный, артрит Органоспецифичные
иммунные заболевания (пернициозная анемия, аутоиммунная гемолитичес-
кая анемия, идиопатическая тромбоцитопеническая
пурпура)
ПВИ — гетерогенная группа
нарушений, характеризующихся гипогаммаглобулине-мией IgG, IgM и IgA. При
этом наблюдается их очень низкий уровень, такой же как при сцепленной с
Х-хромосомой (Брутона) агаммаглобулинемии. Признаки, позволяющие различить
эти заболевания, включают равное распределение среди обоих полов, более
позднее начало симптоматики и наличие циркулирующих В-клеток. Хотя лежащие
в их основе иммунологические нарушения гетерогенны, у большинства пациентов
обнаруживают первичный В-клеточный дефект, выражающийся в неспособности
трансформироваться в иммуносекретирующую клетку плазмы. Как при избирательном
иммунодефиците IgA, в большинстве случаев ПВИ имеет генетически обусловленное
заболевание, однако у некоторых больных ПВИ может развиться как приобретенное
состояние, вторичное к вирусной инфекции или побочному действию лекарственных
препаратов.
Частота септического артрита,
вызываемого Staphylococcus aureus (внутриклеточной бактерией с клеточной
стенкой) и Mycoplasma, увеличивается с повышением распространенности
ПВИ. Грибки и микобактерии тоже должны рассматриваться как потенциальные
возбудители, так как клеточный иммунодефицит иногда встречается при ПВИ.
Наличие аутоантител при отсутствии
клинически выраженного иммунологичес-кого заболевания при ПВИ встречается
реже, чем при избирательном иммунодефиците IgA. Тем не менее аутоиммунные
заболевания при ПВИ — не редкость. Описан полиартрит, при котором не удалось
обнаружить инфекционный агент, несмотря на тщательную диагностику. Характерным
для синдрома является поражение крупных и средних суставов, в то же время
мелкие суставы кисти и стопы остаются интактны-ми. Ревматоидные узелки,
эрозии, значительная деструкция суставного хряща, как правило, отсутствуют.
Эта форма артропатии часто хорошо лечится в/в введением гаммаглобулина.
14. Какие механизмы ответственны
за возникновение асептического артрита и других аутоиммунных процессов
при синдромах первичного иммунодефицита?
Механизмы неизвестны. Существуют
следующие гипотезы:
1. Асептический артрит при
синдромах первичного иммунодефицита может быть обусловлен редким инфекционным
агентом, который не удается обнаружить существующими методами.
2. Отсутствие секреторного
IgA может приводить к:
— чрезмерной абсорбции антигенов
из кишечника и последующему формированию иммунных комплексов (при всех
заболеваниях, кроме сцепленной с Х-хромосомой агаммаглобулинемии) и болезни
иммунных комплексов; затем иммунные комплексы способствуют образованию
ревматоидного фактора;
— чрезмерной абсорбции суперантигенов
из кишечника, которая сопровождается активацией Т-клеток, имеющих особые
Vp-участки на рецепторах; реакция одного из этих клонов против собственных
антигенов выражается в виде аутоиммунного состояния;
— чрезмерной абсорбции особых
антигенов из кишечника, ведущей к возникновению аутоиммунного процесса
в результате молекулярной мимикрии.
3. Сосуществование первичного
иммунодефицита и аутоиммунного процесса можно рассматривать в большей степени
как случайность, чем как закономерность. Обычные HLA-расширенные гашютипы,
обнаруживаемые при избирательном иммунодефиците IgA и ПВИ, также часто
присутствуют при аутоиммунных заболеваниях, таких как СКВ, сахарный диабет
I типа и ту asthenia grams.
15. Каковы принципы лечения
сцепленной с Х-хромосомой агаммаглобулинемии, простого вариабельного иммунодефицита
и избирательного иммунодефицита IgA?
Сцепленная с Х-хромосомой
агаммаглобулинемия: в/в введение иммуноглобулина
и мощная антибиотикотерапия
бактериальной инфекции. Внутривенно иммуноглобулин обычно назначают в дозе
200-600 мг/кг 1 раз в месяц.
ПВИ: гаммаглобулин вводят
в/в тем пациентам с ПВИ, у которых имеются низкая концентрация IgG и рецидивирующие
инфекции. Иногда у больных ПВИ полностью отсутствуют IgA и одновременно
наличествуют aHTH-IgA-антитела, что создает риск анафилаксии при введении
внутривенно иммуноглобулина.
Избирательный иммунодефицит
IgA: длительное лечение активной бактериальной инфекции антибиотиками.
Иммуноглобулин внутривенно назначать не следует, так как многие
пациенты имеют аутоантитела против IgA, включая IgE-aHTH-IgA, и введение
препарата может привести к тяжелой, иногда фатальной, анафилаксии. Лучше
всего больным вводить препараты крови, полученные от других пациентов с
дефицитом IgA.
16. Как проводят скрининговое
исследование на наличие гомозиготного дефицита комплемента у пациента с
ревматическим заболеванием?
Гомозиготный дефицит определенных
компонентов комплемента связан с большим количеством ревматологических
синдромов. С помощью теста на общий гемо-литический комплемент (СН5о)
исследуют целостность классического пути активации комплемента. Сыворотка
пациента добавляется к стандартизированной суспензии овечьих эритроцитов,
покрытых антителами кролика. Этот "иммунный комплекс" вызывает активацию
классического пути и лизис эритроцитов. СН5о — это разведение
плазмы, при котором отмечается лизис 50 % эритроцитов барана. Данный анализ
— идеальный недорогостоящий метод, так как определенные дефициты комплемента,
сопровождающиеся ревматологическими синдромами, проходят чаще по классическому
пути активации, нежели по альтернативному. Гомозиготный дефицит конкретных
компонентов комплемента по классическому пути выражается в СН50
равной 0; содержание каждого компонента комплемента определяется иммунотестами.
17. Каковы ревматологические
проявления состояний гомозиготного дефицита комплемента?
Дефициты "ранних" компонентов
классического пути (С1, С4, С2) связаны с болезнями иммунных комплексов,
особенно СКВ. Это, вероятно, происходит из-за неспособности иммунных комплексов
находиться в растворенном состоянии и неспособности организма удалять эти
комплексы из крови. Наиболее часто встречается дефицит С2-компонента комплемента.
Дефицит компонентов мембраноатакующего
комплекса (С5-С9) связан с рецидивирующей инфекцией, вызываемой Neisseria,
Neisseria meningitidis и Neisseria gonorrhoeae. Больных с рецидивирующей
нейссериальной инфекцией, особенно системной, необходимо обследовать на
наличие дефицита комплемента.
Избранная литература
Buckley R. H. Specific immunodeficiency
diseases, excluding AIDS. In: Kelley W. N.. Harris E. D. Jr, Ruddy S.,
Sledge С. В. (eds). Textbook of Rheumatology, 4th ed. Philadelphia, W.
B. Saunders, 1993,1264-1282.
Lee A. H., Levinson A. I.,
Schumacher H. R. Jr. Hypogammaglobulinemia and rheumatic disease. Semin.
Arthritis Rheum., 22: 252,1993.
Liblau R. S., Bach J.-F.
Selective IgA deficiency and autoimmunity. Int. Arch. Allergy Immunol.,
99:16,1992.
Ruddy S. Complement deficiencies
and rheumatic diseases. In: Kelley W. N., Harris E. D. Jr, Ruddy S., Sledge
C. B. (eds). Textbook of Rheumatology, 4th ed. Philadelphia, W. B. Saun-ders,
1993,1283-1289.
Waldmann T. A., Nelson D.
L. Inherited immunodeficiencies. In: Frank M. M, Austen K. F., Claman H.
N., Unanue E. R. (eds). Samter's Immunologic Diseases, 5th ed. Boston,
Little, Brown & Company, 1995,387-429.
ГЛАВА 63. ДИСПЛАЗИИ
КОСТЕЙ И СУСТАВОВ
Edmund H. Hornstein, D.O.
1. Что такое дисплазия
костей или суставов?
Дисплазия — это термин, обозначающий
аномальный рост, формирование. Применительно к костной системе им определяется
группа состояний, при которых имеют место аномалии роста эпифиза, метафиза,
физиса или диафиза развивающейся кости. Эти аномалии могут давать соответствующую
симптоматику и даже приводить к гибели больного, но они также встречаются
как неожиданные находки на рентгенограмме. В широком смысле слова эти нарушения
называются остеохондродисплази-ями. Большинство синдромов достаточно
редки и выходят за пределы врачебного интереса в большинстве клинических
ситуаций.
2. Почему следует рассматривать
дисплазии костей и суставов в аспекте ревматологии?
• Диспластические синдромы
могут проявляться нарушением функции костно-мышечной системы или болью,
которые напоминают черты других ревматоло-гических синдромов.
• Ранняя диагностика и своевременное
начало лечения некоторых дисплазии могут предупредить или уменьшить в дальнейшем
инвалидизацию и боли.
• Многие хондродисплазии
носят наследственный характер, поэтому точный диагноз позволяет проводить
соответствующую генетическую консультацию.
3. Как классифицировать
остеохондродисплазии?
Существует много классификаций
данных синдромов. С практической стороны полезно группировать заболевания
в зависимости от того, в каком отделе кости отмечаются выраженные нарушения
ее развития. Для облегчения запоминания такой наиболее простой группировки
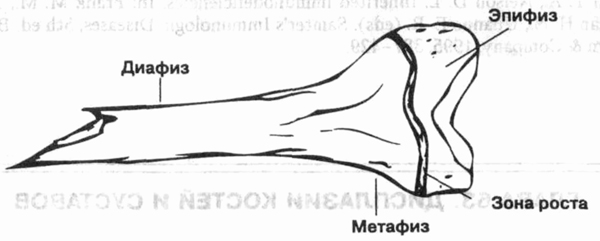
существует мнемонический ключ EMPD (empty — пустой):
E (Epiphysis) — дисплазия
эпифиза. Эпифиз является областью вторичной осси-фикации и концевой частью
длинной трубчатой кости. Для правильного формирования суставной поверхности
необходимо нормальное развитие эпифиза.
M (Metaphysis) — дисплазия
метафиза. Метафиз более широкая, чем эпифиз, часть трубчатой кости между
диафизом и физисом.
Р (Physis) — дисплазия физиса.
Физис, или эпифизарная хрящевая пластинка, отделяет метафиз от эпифиза
во время роста кости. Это главная область кости, отвечающая за ее рост
в длину.
D (Diaphysis) — дисплазия
диафиза. Диафиз — это основная часть длинной трубчатой кости, ее ось. Он
состоит из губчатой и кортикальной частей и покрыт надкостницей.
4. Каковы отличительные
признаки дисплазии эпифиза?
Дисплазия эпифиза характеризуется
аномальной оссификацией растущего эпифиза. Конечные морфологические аномалии
центров окостенения используются для определения разных подтипов этой категории.
Наиболее важны с точки зрения ревматолога дисплазии эпифиза — это множественная
дисплазия эпифиза и спондилоэпифи-зальная дисплазия.
5. Как клинически проявляется
множественная дисплазия эпифиза?
Обычно пациенты жалуются
на симметричные боли в тазобедренных, коленных, лучезапястных и голеностопных
суставах. Часто этим болям сопутствуют боли в спине. Отмечается ограничение
движений в пораженных суставах. На рентгенограмме находят беспорядочные,
ровные по форме и маленькие центры окостенения в детском возрасте и деформированную
суставную поверхность после закрытия метафизарной пластинки (физиса). Отмечается
выраженное поражение длинных костей верхних и нижних конечностей. Позвонки
обычно уплощены (платиспон-дилия), с неровными краями. Взрослые больные
небольшого роста (пропорционально тяжести заболевания). В итоге развивается
инвалидизирующий дегенеративный артрит. Симптоматика появляется в доподростковом
периоде и может быть неявной до юношеского возраста. Она зависит от тяжести
деформации эпифизов костей.
6. Какие состояния могут
быть ошибочно приняты за множественную дисплазию эпифиза?
Воспалительный артрит.
Боль
и симметричное поражение суставов иногда ошибочно воспринимаются за артрит
инфекционной этиологии. Отсутствие признаков воспаления — достаточный повод
для исключения этого заболевания.
Гипотиреоз. Скрытый
гипотиреоз приводит к постепенному развитию аномалий скелета, которые могут
сильно напоминать таковые при некоторых врожденных дис-плазиях эпифиза.
Необходимо каждый раз, когда
предполагается диагноз дисплазии эпифиза, проверять функцию щитовидной
железы.
Ювенильные остеохондрозы.
Эти
заболевания, включая болезнь Легга-Каль-ве-Пертеса (Legg-Calve-Perthes),
рентгенологически похожи на дисплазию, однако при них обычно поражается
один сустав.
7. Опишите типичную рентгенологическую
картину спондилоэпифизальной дисплазий.
Спондилоэпифизальная дисплазия
— группа заболеваний, объединенных сходной рентгенологической картиной:
выраженная платиспондилия (короткие уплощенные позвонки), связанная с нарушением
оссификации эпифизов. Аномалии позвоночника значительные, и постановка
диагноза облегчается наличием нарушения роста позвоночного столба.
8. Почему позднюю спондилоэпифизальную
дисплазию и позднюю спондилоэпи-физальную дисплазию с прогрессирующей артропатией
иногда путают с юве-нильным ревматоидным артритом?
Оба эти заболевания являются
сцепленными с Х-хромосомой рецессивными формами патологии, при которых
отмечается увеличение концевых частей длинных трубчатых костей верхних
конечностей, что при обычном осмотре может быть ошибочно принято за проявления
ревматоидного артрита.
9. Какие аномалии характерны
для дисплазий метафизов?
Эти формы дисплазий развиваются
в результате либо нарушения формирования, либо разрушения губчатой части
кости в процессе ее развития. С точки зрения ревматолога важными дисплазиями
этой категории являются гипофосфатазии и кранио-метафизарные дисплазий.
10. Какова первичная дифференциальная
диагностика при гипофосфатазиях?
Гипофосфатазии могут напоминать
рахит в детском возрасте и остеомаляцию у взрослых. Трудноуловимые рентгенологические
признаки позволяют обнаружить разницу, но диагноз гипофосфатазии в конечном
счете строится на более основательных данных: выявляются исключительно
низкая активность щелочной фосфатазы в сыворотке крови наряду с высоким
содержанием в моче и сыворотке крови фосфорилэтаноламина. Предполагать
наличие гипофосфатазии необходимо при любом случае рахита или остеомаляции.
11. Перечислите основные
сходные признаки различных форм краниометафизар-ной дисплазии.
Суставная боль, мышечная
слабость, сколиоз, патологические переломы, genu valga, колбовидная
деформация длинных костей Эрленмейера (Erlenmeyer). Аномалии черепа включают:
выраженность надбровных дуг, широкий плоский нос, гипертелоризм, аномалию
зубного прикуса и склерозированные толстые кости свода черепа.
12. Одним из наиболее частых
синдромов остеохондродисплазии является синдром, который считается дисплазией
физиса и приводит к карликовости. Назовите этот синдром.
Ахондроплазия. Эта дисплазия
физиса наследуется по аутосомно-доминантному типу, хотя, возможно, в большинстве
случаев имеют место спонтанные мутации. Она рассматривается как диспропорциональная
карликовость с ризомелией (про-ксимальная часть конечности короче дистальной)
укороченных конечностей, макроцефалией с выраженными лобными буграми и
некоторой гипоплазией лицевого отдела. Обычно отмечают увеличенный поясничный
лордоз и сгибательные контрактуры в локтевых и тазобедренных суставах.
Интеллект нормальный. Средний рост взрослого мужчины — 130 см, женщины
— 122,5 см. Ревматологические осложнения могут быть обусловлены сужением
позвоночного канала и симптомами сдавления спинного мозга, а также дряблостью
связочного аппарата коленного сустава, что вызывает жалобы пациента на
боль и приводит к раннему развитию дегенеративного процесса.
13. Где выявляются аномалии
формирования кости при диафизарных дисплазиях?
Дисплазии диафизов происходят
из-за нарушения формирования эндостальной или периостальной части кости.
Эти дисплазии можно подразделить на гиперплазии и гипоплазии. Несовершенный
остеогенез считается гипопластической диафизарной дисплазией (глава 59).
15. Другая диафизарная дисплазия
имеет клинические и рентгенографические признаки, сходные с гипертрофической
легочной артропатией, включая пальцы в виде барабанных палочек, болезненные
отечные суставы, увеличение периостальной части кости, но при этой дисплазии
отмечают толстую морщинистую, как у слона, кожу. Назовите заболевание.
Пахидермопериостоз. Литературный
перевод термина описывает все основные проявления этого страдания.
14. Мужчина двадцати одного
года жалуется на боли в нижней части ног и отеки, которые постепенно сливаются.
Проведено рентгенологическое обследование (см. рисунок). Что это за заболевание?
Мелореостоз — ненаследственная
идиопатическая диафизарная дисплазия. Пациент жалуется на суставные боли,
которые начинаются обычно в позднем детском возрасте или юношестве. Другие
проявления этого редкого заболевания включают: снижение амплитуды движений
в суставах, их контрактуру или анкилоз, нарушения роста, деформацию стоп,
дистрофию кожи, мышц, изменения мягких тканей над пораженной костью. На
рентгенограмме выявляются характерные плотные, волнообразные разрастания
периостальной части кости, которые описываются как воск, стекающий по свече
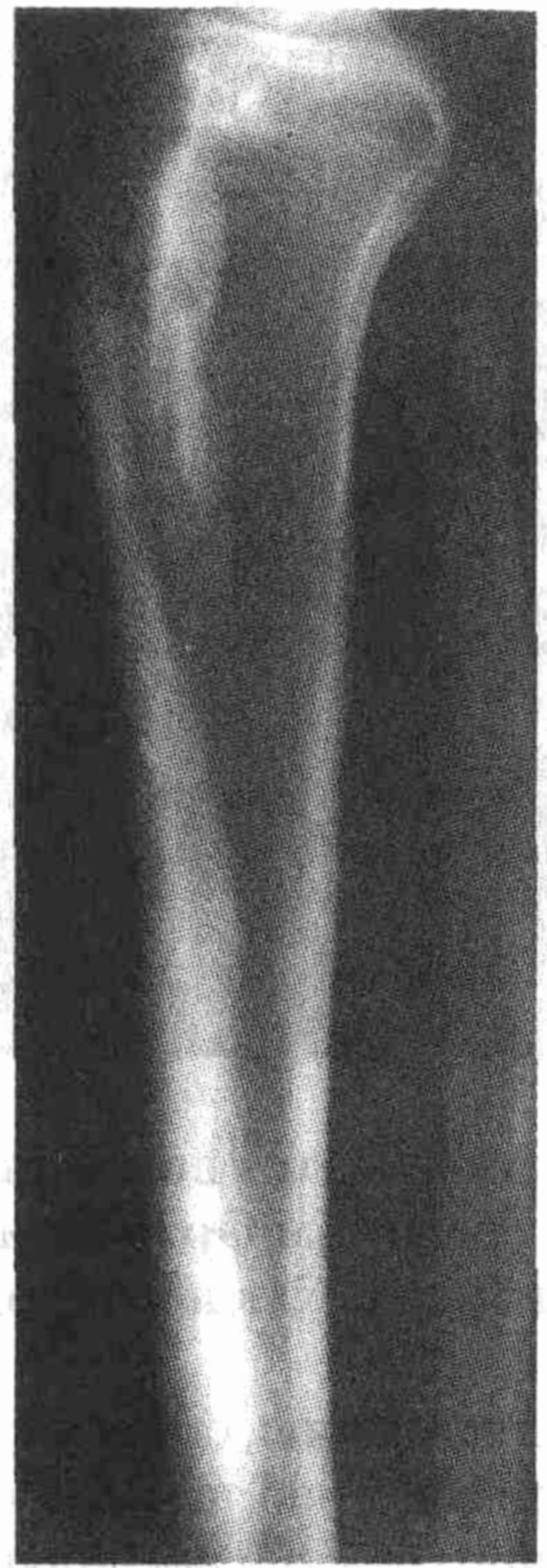
16. Мальчик 15 лет при
осмотре жалуется на боли в верхней части спины, травму грудного отдела
позвоночника отрицает. Боль усиливается при движении, в покое стихает и
не связана с утренней скованностью. При осмотре выявлены только тенденция
к увеличению грудного кифоза с небольшой болезненностью при пальпации в
нижней части грудной клетки сзади по средней линии и небольшой спазм паравертебральных
мышц. По лабораторным данным, отмечается нормальная СОЭ, нет изменений
биохимического и клинического анализов крови. Данные, приведенные в описании
рентгенограммы, указывают на болезнь Шейерманна. В чем здесь "загвоздка"?
Позвоночный остеохондрит,
или болезнь Шейерманна (Sheuermann),— возрастная аномалия оссификации замыкательных
пластинок позвонков, наболее часто наблюдаемая в грудном отделе позвоночника,
но также в пояснично-грудном и поясничном отделах. Она встречается у подростков
и в 60 % случаев симптоматична, но может обнаруживаться как находка при
рентгенологическом исследовании, выполненном по другой причине. На рентгенограмме
обращают на себя внимание клиновидные изменения нескольких позвонков с
клином, обращенным кпереди, с узлами Шморля (грыжи Шморля) и неровностью
их замыкательных пластинок. Патогенез болезни неясен, но существует наследственная
слабость замыкательных пластинок, обнаруживаемая у больных. Эта слабость,
как считают, приводит к просачиванию материала межпозвонкового диска в
тело позвонка, что, в свою очередь, приводит к нарушению роста и описанной
выше рентгенологической картине. Лечение обычно носит симптоматический
характер и направлено на ограничение увеличения грудного кифоза. Иногда
требуется хирургическое вмешательство.
17. У новорожденной девочки
отмечается воспроизводимый щелчок при сгибании и отведении правой ножки
(симптом Ортолани). Подозревается наличие у ребенка врожденного вывиха
бедра. Как подтвердить диагноз? Что сообщить родителям?
Врожденный вывих бедра, или
дисплазию, обнаруживают сразу после рождения при физикальном осмотре и
проведении проб, таких как симптом Ортолани (Ortolani), и провоцируя вывих
нестабильного бедра и последующее изменение его формы (симптом Барлоу [Barlow]).
Рентгенограмма в переднезадней проекции довольно трудна для интерпретации
в первые недели жизни, но другие исследования, как то ультразвуковое исследование,
компьютерная томография и МРИ, более чувствительны.
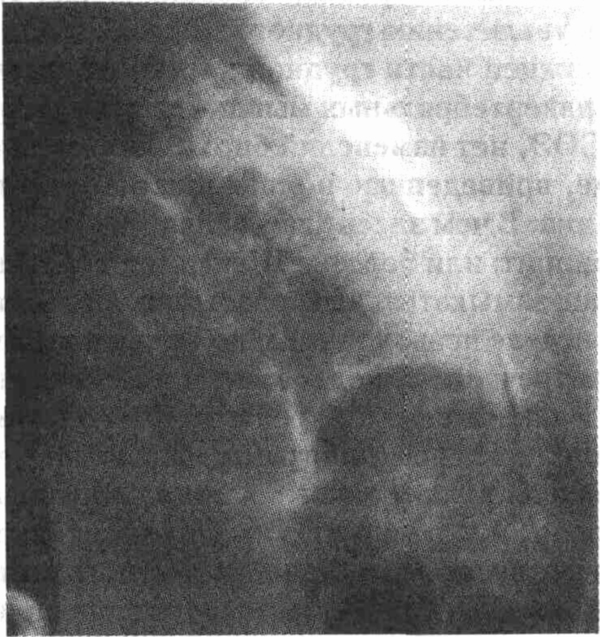
Рентгенограмма взрослого человека с
врожденной дисплазией тазобедренного сустава, которому не проводилось лечение
в детстве. Отмечаются выраженные дегенеративна изменения, мрл^ця вертлужная
впадина и деформированная головка бедренной кости вдшиМ вкофпм слон
Врожденная дисплазия тазобедренного
сустава имеет хороший прогноз, если она выявлена сразу после рождения.
Лечение включает фиксацию нижних конечностей в положении абдукции, что
позволяет головке бедра полностью занимать мелкую ацетабулярную впадину.
Если диагноз поставлен поздно, то лечение заболевания сложно и может потребоваться
ортопедическое вмешательство. При отсутствии лечения возникает ранний остеоартрит,
и тогда уже требуется полная пластика головки бедренной кости.
Избранная литература
Altman R. D, Tenenbaum J.
Hypertrophic osteoarthropathy. In: Kelley W. K., Harris E. D., Ruddy S.,
Sledge C. B. (eds). Textbook of Rheumatology, 4th ed. Philadelphia, W.
B. Saun-ders, 1993,1545-1563.
Borenstem D. G., Wiesel S.
W. Vertebral osteochondntis. In: Borenstem D. G., Wiesel S. W., Boden S.
D. (eds). Low Back Pain: Medical Diagnosis and Comprehensive Management,
Philadelphia, W. B. Saunders, 1989, 227-229.
Clark R N. Congenital dysplasias
and dwarfism. Pediatr. Rev., 12(5): 149-159,1990.
Horton W. A., Hall J. G.,
Scott C. I. et al. Growth curves for height for diastrophic dysplasia.
Spondyloepiphyseal dysplasia congenita and pseudoachondroplasia. Am. J.
Dis. Child., 136: 316-319,1982.
Kozlowski K. Metaphyseal
and spondylometaphyseal chondrodysplasias. Clin. Orthop., 114:83-93,1976.
Lateur L. M. Bone and joint
dysplasias. In: Klippel J. H., Dieppe P. A. (eds). Slide Atlas of Rheumatology.
St Louis, Mosby, 1994, sect 7, unit 3,45.3-45.10.
Lowe Т G. Scheuermann disease.
J. Bone Joint Surg., 72A: 940-945,1990.
Pyentz R E. Heritable disorders
of connective tissue. In: Schumacher H. R. (ed.) Primer on the Rheumatic
Diseases, 10th ed., Atlanta, Arthritis Foundation, 1993, 249-255.
Scott C. I. Achondroplastic
and hypochondroplastic dwarfism. Clm. Orthop., 114: 18-30,1976. Spranger
J. The epiphyseal dysplasias. Clm. Orthop., 114:47-59,1976.
Stemberg M. E., Stemberg
D. R. Osteonecrosis. In: Kelley W. K., Harris E. D., Ruddy S., Sledge C.
B. (eds). Textbook of Rheumatology, 4th ed. Philadelphia, W. B. Saunders.
1993, 1628-1650.
Townsend D. J , Tolo V. Т
Congenital dislocation of the hip. Curr. Opm. Rheumatol., 6: 183-186,1994
Wenger D. R. Legg-Perthes
disease. In. Klippel J. H., Dieppe P. A. (eds). Slide Atlas of Rheumatology.
St. Louis, Mosby, 1994, sect 7, unit 3, 42.1-42.4.